Command Palette
Search for a command to run...
Protein Structure prediction/function annotation/interaction recognition/on-demand Design, China Ocean University Zhang Shugang Team Directly Hits the Core Tasks of Protein Intelligent Computing
As the main bearer of life activities, proteins play a key role in human physiological functions. However, traditional research faces challenges such as high cost of structural analysis, serious lag in functional annotation, and low efficiency in the design of new proteins. In recent years, the demand for the analysis of complex properties of proteins in life sciences has become increasingly urgent. The breakthrough development of technologies such as big data, deep learning, and multimodal computing has provided a new development opportunity for the construction of a protein intelligent computing system. The construction of a protein intelligent computing system has enabled proteins to achieve remarkable results in the fields of large-scale functional annotation, interaction prediction, and three-dimensional structure modeling, providing a new technical path for drug discovery and life system simulation.
At the 2025 Beijing Zhiyuan Conference, Associate Professor Zhang Shugang from the School of Computer Science at Ocean University of China spoke on the topic of "Construction and Application of Protein Intelligent Computing System" in the "AI+Science & Engineering & Medicine" forum.Starting from the core value of protein intelligent computing system, the paper systematically explains the technical breakthroughs of four core tasks: protein structure prediction, functional annotation, interaction recognition and new design.The team's relevant research results were highlighted.

HyperAI has compiled and summarized Associate Professor Zhang Shugang’s in-depth sharing without violating the original intention. The following is the transcript of the speech.
Overview of protein intelligent computing system: AI-driven revolution in life sciences
In life science research, the importance of protein is self-evident. It is not only an enzyme that catalyzes biochemical reactions, but also a messenger that transmits signals, constitutes the structural basis of the body, and is the "weapon" of the immune system to resist foreign enemies. However, traditional research methods seem to be powerless when faced with the complex characteristics of proteins. Problems such as high cost of structural analysis, serious lag in functional annotation, and low success rate of protein design have become important challenges.
The introduction of AI technology has completely reversed this situation. In 2024, the Nobel Prize in Chemistry was awarded for breakthroughs in AI protein structure prediction and design, which undoubtedly once again fully demonstrated the important position of AI in protein research.Protein intelligent computing achieves efficient simulation and prediction of complex protein properties by building data-driven algorithm models.It also provides new ideas and research paradigms for addressing the above challenges and opens a new era for life science research.
Breakthrough in the core task of protein intelligent computing
The core issues of protein intelligent computing are the following four categories:
Can protein structure be predicted from scratch?
From the Levinthal Paradox to the Subversion of AlphaFold
Taking protein folding as an example, a protein with 100 residues may have up to 10 possible conformations.200 If there is a random search, the time required is much longer than the age of the universe (13.8 billion years), which is the famous Levinthal paradox. However, actual protein folding can be completed within milliseconds to minutes, which suggests that there is a specific folding path.
In 2018, the first generation of AlphaFold model attempted to solve the problem using deep learning methods, using the residual convolution module to predict the distance and torsion angle of amino acid pairs.In CASP13, it led other competitors by a significant margin, accurately predicting 25 protein structures.The second place only predicted 3 correctly.
In 2021, the second-generation model achieved a qualitative leap. AlphaFold2 used HMMER and HH-suite to perform multiple sequence alignment and template search.Through 48 Evoformer modules and 8 Structure modules, atomic-precision protein structure prediction is achieved.A database containing about 214 million protein monomer predictions was released. The average error between the predicted structure and the electron microscopy analysis results does not exceed one atom width, reaching the "Highly Accurate" standard.
In 2024, the third-generation model will further achieve full prediction of the in vivo protein interaction structure. AlphaFold3 has achieved a qualitative leap. It can not only predict protein structure,It can also predict the structure of complexes composed of proteins, nucleic acids, small molecules, ions and all other life molecules.It covers almost all molecular types in the PDB database, providing a powerful tool for understanding cell functions and disease treatment.
Can protein functions be automatically annotated: a breakthrough in multi-source data fusion
Due to the forward-looking progress of AlphaFold3 in the field of protein prediction, our team decided to shift the focus of research to the field of protein function annotation and interaction analysis. Currently, among the 250 million protein sequences in the world, only 0.5% has completed accurate functional annotation. The traditional model that relies on manual analysis by biological experts has been unable to cope with the challenge of massive data. Therefore, using deep learning to achieve large-scale batch annotation has become a key breakthrough.
Our exploration in this field began in 2022. Aiming at the industry pain point that electron microscopy structural data, which deep learning relies on, is scarce and costly,We innovatively propose to use the virtual structure data predicted by AlphaFold2 in model training.This strategy, similar to "data enhancement", significantly expanded the scale of training data - from the 5 million samples that traditional electron microscopes can provide to a theoretically large prediction data pool of hundreds of millions. Experimental verification shows that the model trained based on prediction data not only outperforms the native version, but can also discover new protein functions that are not identified by traditional methods.
Paper title:Enhancing Protein Function Prediction Performance by Utilizing AlphaFold-Predicted Protein Structures
Paper address:
https://pubs.acs.org/doi/10.1021/acs.jcim.2c00885
In terms of technological innovation,To address the problem of insufficient protein structure information mining, our team proposed a protein function prediction method based on self-supervised graph attention.By encoding the correlation information of residues within the protein molecule and making full use of the distance information between residues as an auxiliary task, the performance of protein function prediction can be improved.
Paper title:SuperEdgeGO: Edge-Supervised Graph Representation Learning for Enhanced Protein Function Prediction (forthcoming publication)
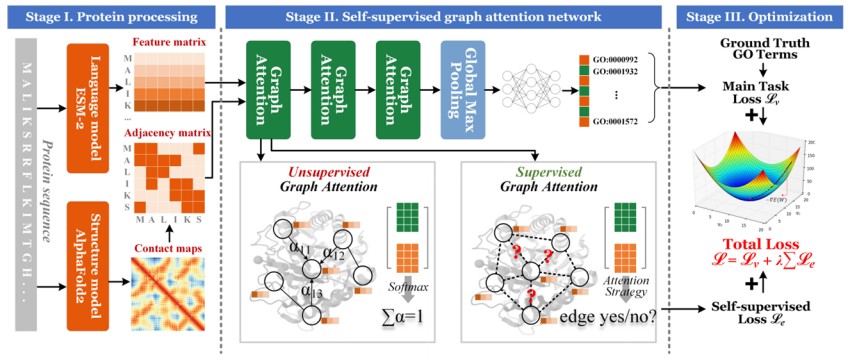
To address the problems of heterogeneous protein features that are difficult to fuse and spatially inconsistent, a protein dual-view construction strategy and feature alignment method were proposed.Based on the complex characteristics of biological proteins with six cross-scale modes (covering sequence, three-dimensional structure, functional domain and other dimensions),The team further proposed a multimodal fusion strategy——Integrate contrastive learning and multi-view analysis methods in the computing field to build a hierarchical feature fusion model. This solution was compared with 20 mainstream baseline methods on 7 data sets, all of which achieved SOTA results, successfully solving the technical problem of performance degradation caused by direct splicing of modalities.
Paper title:Annotating protein functions via fusing multiple biological modalities
Paper address:https://www.nature.com/articles/s42003-024-07411-y
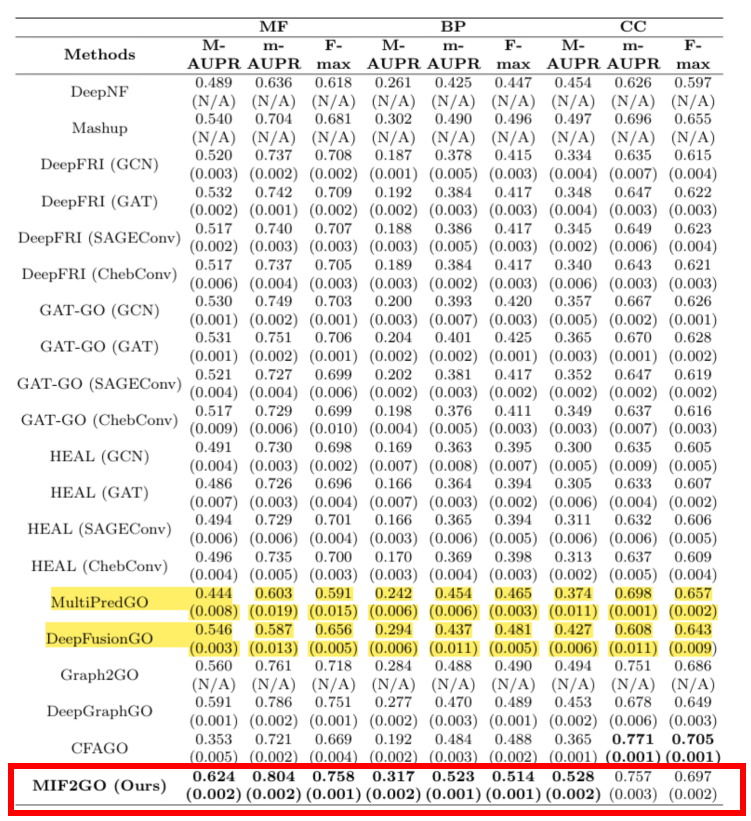
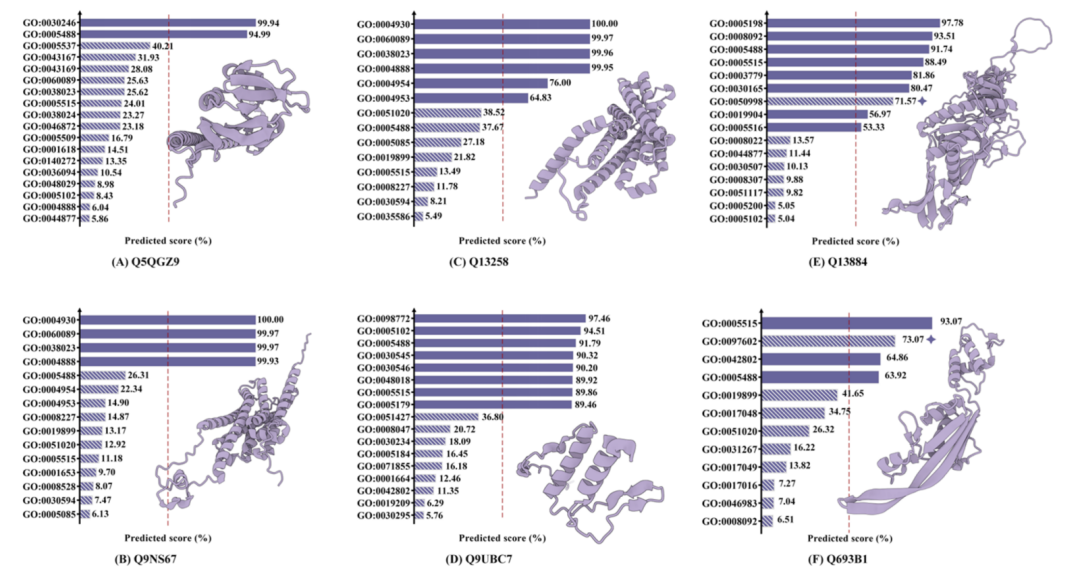
In addition, in the study of the interpretability of functional prediction,The model also demonstrated an excellent ability to accurately identify more than 10 protein functions from thousands of GoTerms annotations.In addition, the team found through literature research that cases where the model predicted errors but gave high confidence have actually been recorded in some studies, indicating that these cases may have been misjudged due to the lag in the data set version. This discovery highlights the potential of AI models in exploring new protein functions.
Can protein interactions be accurately identified? Self-developed models enable efficient predictions
In the field of drug development, the precise docking of proteins as human targets is the key to drug efficacy, and AI technology has shown important value in this process. Although AlphaFold3 has performed well in the field of protein structure prediction, there are obvious limitations in practical applications: its free version only supports 20 visits per day, covers about 15-20 types of molecules, and it is extremely difficult to apply for commercial use rights, which prompted the team to develop its own model.
Based on this problem, the team focused on the following tasks:
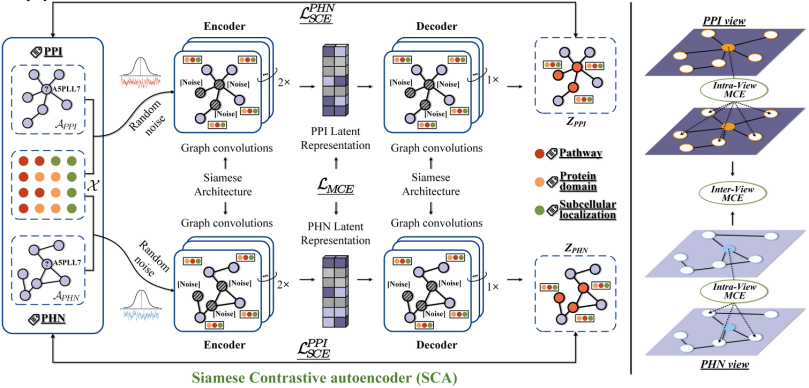
First, we aimed at the problems of poor synergistic interactions in existing protein interaction prediction methods.A twin learning model is introduced in the encoder to enhance the collaborative consistency of protein representation, and a collaborative learning framework with protein interaction collaborative mechanism and task collaborative mechanism is proposed.The team used interactive attention and multi-task learning methods to achieve interactive predictions of protein-nucleic acid, protein-protein, and protein-small molecule.
The team also integrated Transformer and graph neural networks in the NLP field and developed modules such as Convformer and Graphormer to achieve remote interactive modeling.The cross-attention mechanism is used to strengthen the fusion of multimodal information. The model shows strong generalization ability in actual scenarios. Taking the prediction of pancreatic cancer signaling pathway as an example, its accuracy rate exceeds 95%, with only 9 pairs of interaction prediction errors.
Paper title:SSPPI: Cross-modality enhanced protein-protein interaction prediction from sequence and structure perspectives (forthcoming publication)
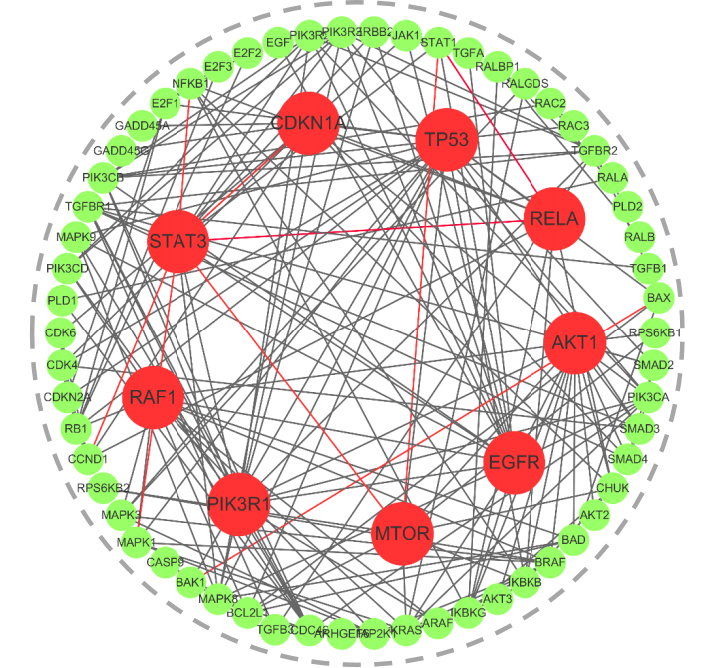
Black line: correct prediction; red line: wrong prediction
In recent research, in addition to cross-scale dimensionality reduction of proteins at the network level, we are also committed to mining protein features. Given that traditional graph models will cause information loss when reducing three-dimensional structural information to two dimensions, we introduced the latest geometric deep learning.A geometric deep learning method based on a hybrid message passing strategy is proposed, and a complete three-dimensional information integration paradigm is constructed.This paradigm aims to solve the irrationality of discarding three-dimensional information in spatial site modeling and provide new research ideas in the field of protein three-dimensional modeling.
Paper title:Geometric Deep Learning for Protein-Ligand Affinity Prediction with Hybrid Message Passing Strategies (forthcoming publication)
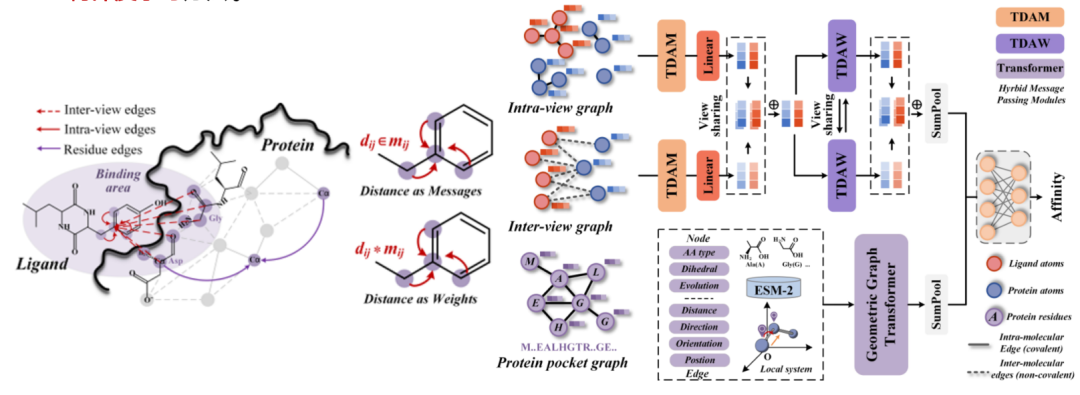
also,We also conducted actual tests on the ACSS2 protein and screened out several candidate compounds from tens of thousands of compounds.The model prediction results indicate that the affinity of the screened compounds can reach the nM level, showing good drug potential; our team cooperated with the team from Qingdao University Medical College to conduct verification, and the docking results were also preliminarily confirmed in the wet experiments conducted recently.
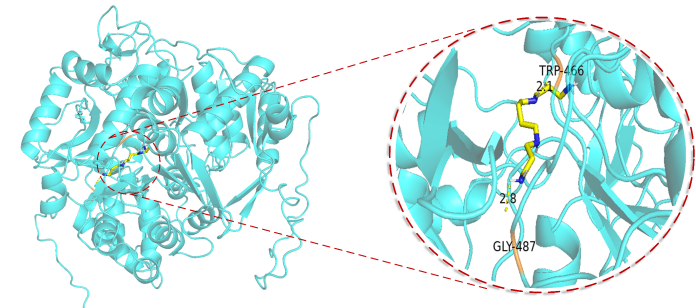
Can new proteins be designed on demand: from inverse problems to innovative applications
Protein design is one of the ultimate goals of protein research and is of great significance for vaccine development, cancer treatment and biomaterial development. However, as an inverse problem of protein folding, protein sequence design also faces challenges such as search space explosion and traditional force field simulation errors.
Facing the core issue of intelligent protein design and optimization, here we take the latest work of Baker's team, last year's Nobel Prize winner, as an example. Snake venom has no specific antidote. Is it possible to design a new type of protein based on a computer? Based on this problem, Baker's team combined its previous ProteinMPNN and RFDiffusion to design a new protein. In addition, his team also carried out the design of specific binding proteins for snake venom toxins, providing a new solution for neutralizing lethal snake venom toxins. The relevant paper was published in the main journal of Nature in early 2025. These research results demonstrate the great potential of AI in the field of protein design, and have taken solid steps towards the "creator"-like goal of "designing new proteins."
Cross-scale computing of complex life systems: full-chain simulation from nanoscale to macroscale
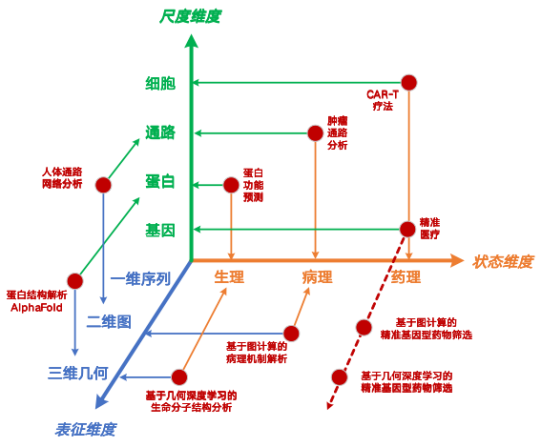
The life system is a complex multi-scale system. From the nanoscopic gene level to the macroscopic cell level, each scale interacts and influences each other. During my visit to Professor Zhang Henggui's research group at the University of Manchester, UK, I conducted research related to the digital heart. After returning to China, I further carried out research on digital cells. Different from the "numerical drive" paradigm such as the digital heart,The team proposed a multi-scale modeling method for microscopic life activities based on a "data-driven" construction approach, and constructed a three-dimensional microscopic computing method system of "representation-state-scale".It covers 36 research points, and currently there are articles or patents accumulated under nearly 1/3 of the methods.
In addition, under the guidance of Professor Wei Zhiqiang,We have newly defined the microscopic life system at four levels of scale.Including the nanoscopic gene level, the "microscopic" protein level, the "mesoscopic" signal pathway level and the "macroscopic" cellular level, the full-chain life system simulation is realized, hoping to achieve full-scale coupling from atoms to the heart.
About Associate Professor Zhang Shugang
Zhang Shugang is an associate professor and master's supervisor at the School of Computer Science at Ocean University of China, a senior member of CCF, a corresponding member of the CCF Bioinformatics Committee, a member of the CAAI Smart Healthcare Committee, a director of the Shandong Bioinformatics Society, and the director of the National Natural Science Foundation of China, the Basic Scientific Research Business Expenses Project of Central Universities, etc. He was selected for the 2020 Shandong Postdoctoral Innovation Talent Support Program.
His main research areas are computational biology and bioinformatics, including ultra-high precision digital heart construction, protein function prediction and design, etc. In recent years, he has published more than 30 papers in international authoritative journals and conferences such as IEEE JBHI, JCIM, and npj Systems Biology and Applications, with over 1,600 citations on Google Scholar.