Command Palette
Search for a command to run...
From 100,000 Possibilities to Successful Synthesis, the Innovative Integration of Global Attention Mechanisms and the AI Model CGformer Facilitate the Development of high-entropy materials.

Artificial intelligence is profoundly reshaping the paradigm of materials science research and development, demonstrating groundbreaking value in accelerating the discovery of new materials and optimizing their performance. Through the deep integration of high-throughput computing and machine learning, the pain points of traditional "trial and error" methods, such as long experimental cycles and high resource consumption, have been effectively addressed.Materials exploration has entered the efficient iterative stage of "computation-driven-experimental verification".However, with the innovation of human technology and lifestyle, the performance requirements for new materials in fields such as new energy and aerospace are becoming increasingly stringent, and the limitations of traditional machine learning methods are gradually becoming apparent, especially in the field of high-entropy material research and development.
High-entropy materials are a new class of materials made from a mixture of multiple principal elements. Through the synergistic action of these elements, high-entropy materials significantly increase the configurational entropy (i.e., disorder) of their atomic arrangement, resulting in superior mechanical, high-temperature, and corrosion-resistant properties compared to traditional materials. These materials hold significant potential for application in energy storage, aerospace, and extreme environment equipment.
Previous approaches, such as Crystal graph convolutional neural networks (CGCNN) and Atomistic line graph neural networks (ALIGNN), all have architectural flaws.Limited by the local information interaction mechanism, it is difficult to model the long-distance atomic synergistic effect and cannot fully capture the global information unique to complex crystal structures, resulting in limited prediction accuracy.At the same time, the inherent properties of high entropy materials also make their research and development face challenges far beyond those of traditional materials.Complex microstructures, scarce high-quality experimental data, and dynamically disordered atomic behavior together constitute key obstacles in the development of high-entropy materials.
In response to tool defects and demand upgrades,The team of Professor Li Jinjin and Professor Huang Fuqiang from the Artificial Intelligence and Microstructure Laboratory (AIMS-Lab) of Shanghai Jiao Tong University has developed a new AI material design model CGformer, which successfully breaks through the limitations of traditional models.This model innovatively combines Graphormer's global attention mechanism with CGCNN, and integrates centrality coding and spatial coding, so that it can not only intuitively describe the material structure through crystal diagrams, but also capture the interactions between long-distance atoms with the help of the "global attention" mechanism, thereby obtaining global information processing capabilities that traditional "only focusing on adjacent atoms" models do not have.
This method provides more comprehensive structural information, enabling more accurate prediction of ion migration within structures, and providing a reliable tool for the research and development of new materials, particularly high-entropy and complex crystalline materials. The research findings were published in the leading journal Matter under the title "CGformer: Transformer-enhanced crystal graph network with global attention for material property prediction."
Research highlights:
* Research and development of CGformer, an AI material design model based on a global attention mechanism, provides a reliable and powerful tool for materials research and development science, helping to accelerate the discovery process of complex crystal structures.
* Compared with CGCNN, CGformer has a 25% reduction in mean absolute error in the study of high-entropy sodium-ion solid electrolytes (HE-NSEs), effectively demonstrating its practicality and advancement.
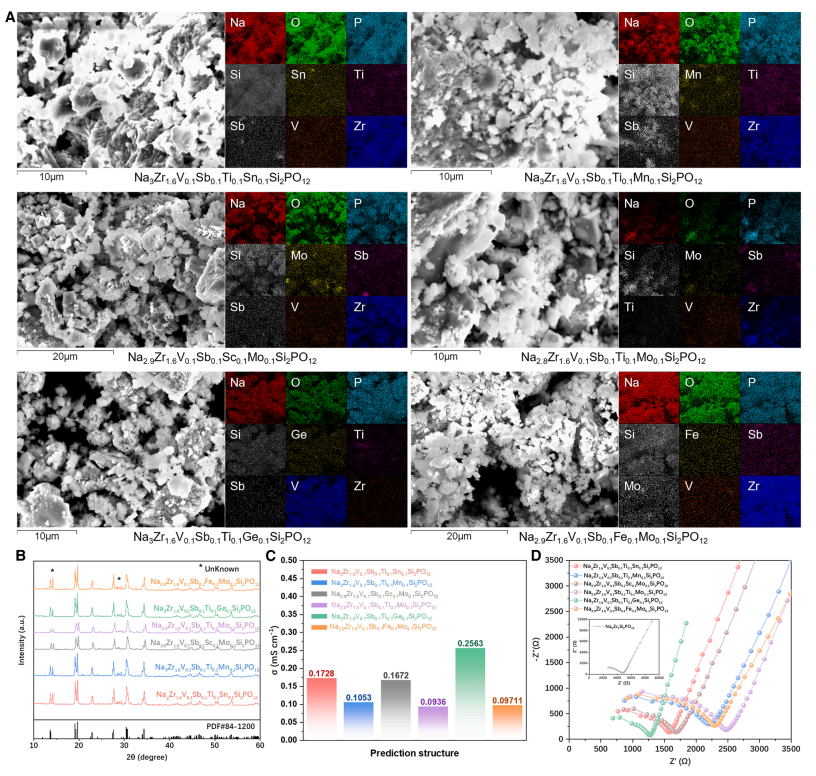
*Screened out 18 possible high-entropy structures from 148,995, and successfully synthesized and verified six high-entropy sodium-ion solid electrolytes (HE-NSEs), with room-temperature sodium-ion conductivity as high as 0.256 mS/cm, demonstrating their practical application value.

Paper address:
https://www.cell.com/matter/abstract/S2590-2385(25)00423-0
Follow the official account and reply "CGformer" to get the complete PDF
More AI frontier papers:
Multi-category datasets improve CGformer model capabilities
The purpose of this study is to address the challenges posed by data scarcity and structural complexity in high-entropy systems through case-based solutions.The research case focuses on new energy electric vehicles and grid energy storage applications, specifically focusing on the performance prediction and screening of high-entropy sodium-ion solid electrolytes. A variety of datasets were constructed and used to support the training, fine-tuning, and experimental verification of the CGformer model, as follows:
Sodium ion diffusion energy barrier (Eb) basic data set:This is the largest known dataset of sodium ion diffusion barriers in high-entropy structures constructed by researchers for this study. It is based on the Crystallographic Analysis by Voronoi Decomposition (CAVD) and Bond Valence Site Energy (BVSE) methods. This dataset is primarily used for pre-training CGformer, enabling the model to learn graph information related to sodium-containing structures, which is then transferred to the calculated high-entropy dataset, laying the foundation for subsequent Eb prediction of high-entropy materials.
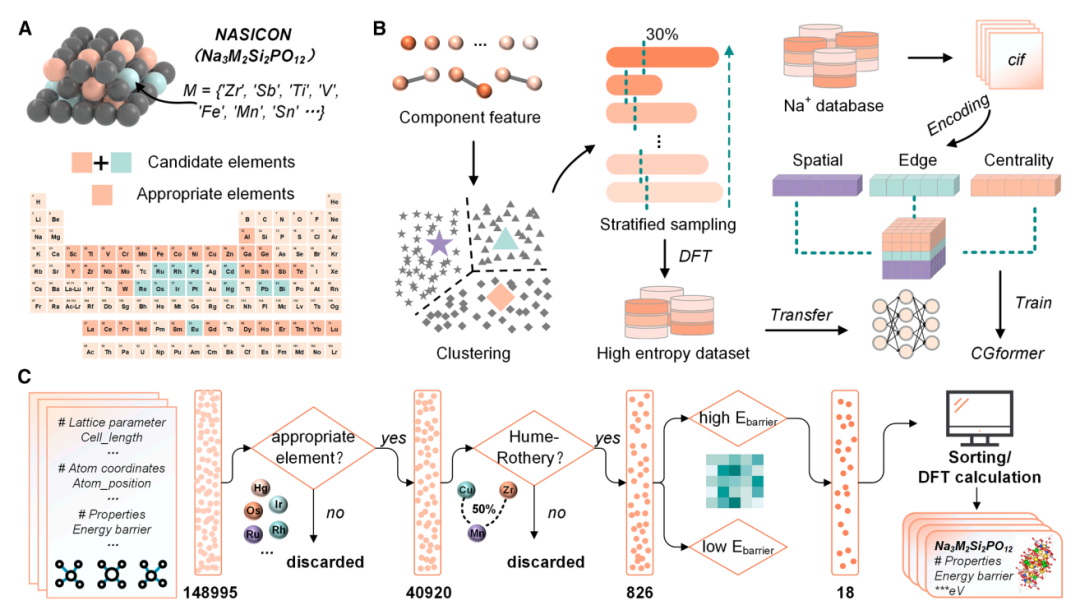
HE-NSEs calculation dataset:Based on Na₃Zr₂Si₂PO₁₂ (as shown above), 45 potential high-entropy doping elements were considered at the Zr site, resulting in an initial chemical space containing 148,995 possible high-entropy structures. Subsequently, through multiple rounds of screening, including the exclusion of unsuitable elements (radioactive, highly toxic, and expensive elements) and constraints on atomic radius differences and charge balance, the chemical space was further narrowed to 826 relatively stable structures. Unsupervised hierarchical clustering was then used to classify these structures into 20 groups. From each group, 30% structures (238 in total) were hierarchically sampled and their Eb values were calculated using density functional theory (DFT). This ultimately formed a dedicated dataset for fine-tuning CGformer, specifically adapting the model to the task of predicting the Eb of sodium ions in high-entropy NASICON structures, improving the model's accuracy in the target scenario.
Thermal stability evaluation dataset:Researchers extracted all sodium-containing structures with energies above the convex hull value (Ehull) from the Materials Project database and compiled them into a dedicated training set. This dataset is primarily used to train a complementary model for evaluating the thermodynamic stability of HE-NSEs. Combined with the Eb predicted by CGformer, this model can be used to screen candidate materials for both high performance and stability.
Innovative fusion architecture enables CGformer's "global perception"
CGformer has made fundamental innovations to address the shortcomings of traditional methods, integrating two advanced technologies to achieve complementary advantages.Its core is to retain the graphical representation ability of crystal structure, and break the limitation of focusing only on local atomic interactions through the global attention mechanism.Specifically, it combines Graphormer's global attention mechanism with CGCNN's crystal graph representation method, while adding key encoding modules to build a new information processing pipeline.
Figure a below shows the crystal diagram encoding process.The process is to convert the real three-dimensional crystal structure into a crystal diagram that can be processed by the model.Atoms in a crystal structure are represented as nodes, and the chemical bonds between atoms are represented as edges. Through this conversion process, researchers were able to extract node and edge features, such as various elemental properties, charge, covalent radius, interatomic distances, bond types, and crystal symmetry information. These features were then combined to obtain the initial input data required by CGformer, ensuring that the chemical and structural information of the crystal was fully preserved.
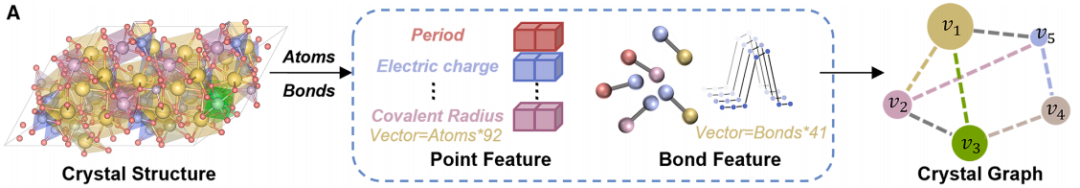
Figure b below shows the network architecture of CGformer, through multi-module collaboration, global information integration and accurate prediction are achieved.First, the input crystal graph undergoes a round of graph convolution operations to generate a simplified graph structure, thereby reducing the computational workload of subsequent network layers and accelerating the training process of CGformer. Then, based on this, the researchers calculate the center code and update the node features of the crystal graph. The center code includes the in-degree and out-degree of each node, which are then integrated into the original node features. Each node is then passed through a multi-head attention module (Multi-head Attention Module), combining variable features and spatial encoding to represent the positional relationship between nodes. The center code converts the average features of adjacent nodes into a sum form, while the spatial encoding enables the self-attention mechanism to distinguish adjacent nodes, promote effective message aggregation, and enhance information connections between different atoms. Finally, the output vector undergoes the "pooling (integration of global features)" and "activation (function operation)" processes to complete the final material property prediction.
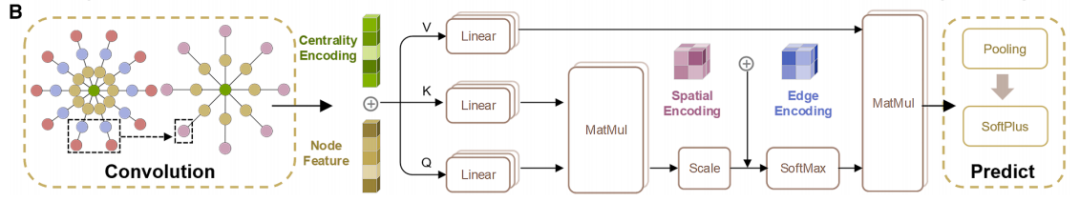
Notably, the multi-head attention module enables each node to "pay attention" to all other nodes in the crystal graph, rather than just adjacent nodes, thus capturing long-range atomic interactions. Furthermore, the addition of center encoding and spatial encoding enables the model to not only identify the chemical properties of atoms but also perceive their "positional importance" and "spatial relationships" within the structure, improving the model's accuracy in characterizing complex crystals.
all in all,Compared with traditional crystal networks, CGformer has achieved a qualitative leap, realizing three major advantages: global vision, information enhancement, and efficiency balance, providing a credible and reliable tool for the discovery and performance optimization of complex high-entropy materials.
CGformer demonstrates powerful performance and highlights its practical guidance value
In order to accurately evaluate the performance and advancement of the CGformer model, the researchers compared it with traditional models such as CGCNN, ALIGNN, and SchNet. The experiment verified the prediction accuracy of CGformer from two stages: "pre-training" and "fine-tuning".
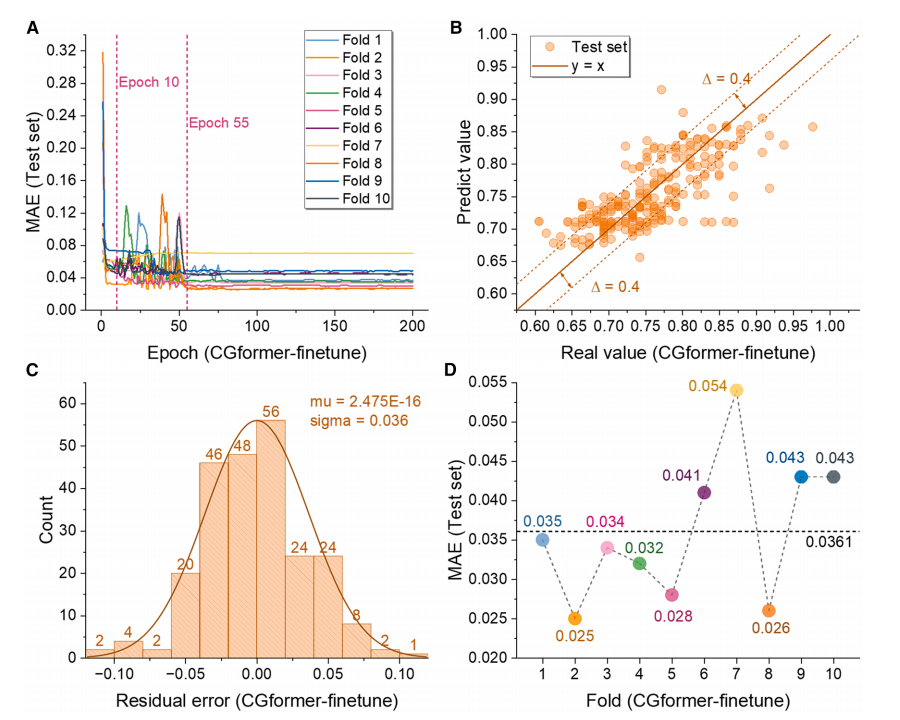
During the pre-training phase (as shown in the figure below), CGformer demonstrated superior stability and prediction accuracy. Its initial error and fluctuation were significantly lower than those of CGCNN. Ten-fold cross-validation (10-fold CV) showed that its training set mean absolute error (MAE) was 0.1703, an improvement of 25.71 TP³T over CGCNN; its test set average MAE was 0.3205, an improvement of nearly 101 TP³T over CGCNN. Comparisons with ALIGNN and SchNet further highlighted CGformer's superior performance.
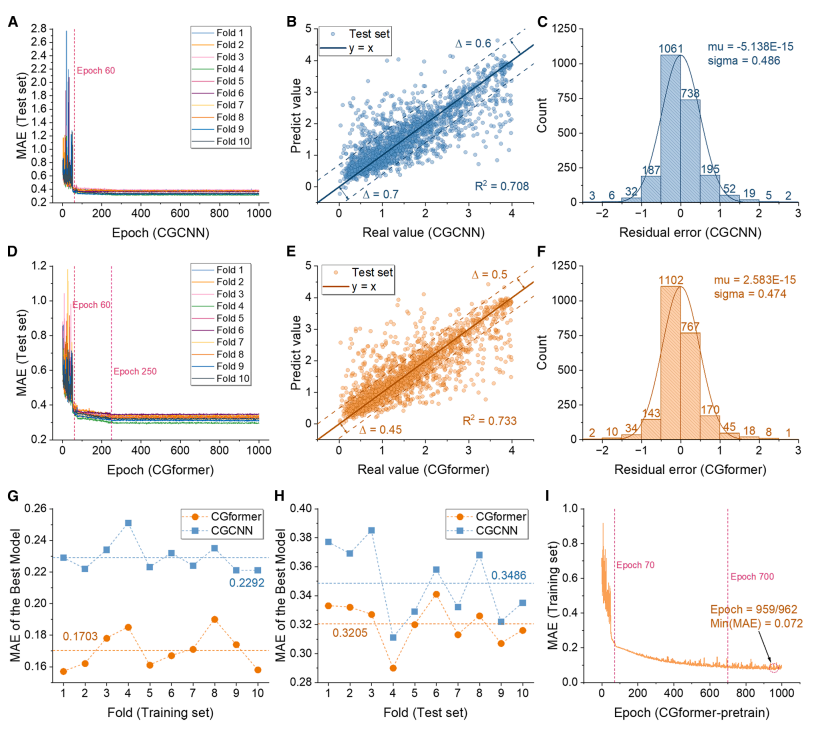
From the fitting results, the predicted value of CGformer deviates less from the true value, the residual is more concentrated near 0, and the residual standard deviation is smaller, which proves that its prediction of sodium ion Eb is more reliable.
During the fine-tuning phase (as shown in the figure below), the MAE of the pre-trained CGformer dropped significantly after about 10 rounds of fine-tuning out of 200 rounds, and the final average MAE of the 10-fold cross-validation was only 0.0361. After fine-tuning, the deviation between the predicted value and the true value was further reduced, and the residuals were mainly concentrated in the range of -0.05 to 0.05, and showed a good normal distribution, demonstrating its extremely high accuracy in predicting the high-entropy system Eb and reflecting its application potential in data-missing scenarios.
Finally, the six optimal HE-NSEs selected by CGformer were synthesized and electrochemically characterized to verify their structure and performance. The results showed that these materials exhibited excellent room-temperature ionic conductivity, with sodium ion conductivity ranging from 0.093 to 0.256 mS/cm at 25°C, significantly higher than that of undoped Na₃Zr₂Si₂PO₁₂.
"Artificial Intelligence + Materials" has become the mainstream of materials science development
"Artificial intelligence + materials" has become a cutting-edge research direction in the current field of materials science. By integrating artificial intelligence technology with material research and development, design and application, it demonstrates the strong development potential and application value of the intersection of the two disciplines. The introduction of CGformer has undoubtedly made a significant contribution to the application of artificial intelligence in the field of materials science. Due to its unique and innovative algorithmic architecture, it solves the key problems in the research and development of high-entropy materials.
CGformer is just the tip of the iceberg in AIMS-Lab's exploration of the fields of artificial intelligence and materials science. As one of the laboratory's main research directions, the interdisciplinary research of artificial intelligence and materials science has long become a vivid footnote of the laboratory and has yielded fruitful results.
Last year, the same team presented their research findings in the leading international journal Energy Storage Materials, titled "Transformer enables ion transport behavior evolution and conductivity regulation for solid electrolytes." The study proposed an artificial intelligence model called T-AIMD, which utilizes a Transformer network architecture. This model significantly reduces computational costs while enabling rapid and accurate prediction of the behavior of any ion in any crystal structure. This approach increases the speed of traditional AIMD simulations by over 100 times, significantly accelerating the process of evaluating material properties.
Paper address:
https://www.sciencedirect.com/science/article/abs/pii/S2405829724003829
Separately, teams from the Technical University of Berlin and the University of Luxembourg in Germany published related research in AIP Publishing, proposing SchNet, a deep learning architecture specifically designed for simulating atomic systems. This architecture utilizes continuous filtering convolutional layers to learn chemically plausible embeddings of atomic types in the periodic table, demonstrating powerful capabilities in predicting various chemical properties of molecules and materials. The paper is titled "SchNet – A deep learning architecture for molecules and materials."
Paper address:
From everyday plastic packaging and metal products to nanomaterials and superconductors in high-end industries, the progress of human civilization is closely linked to the development of materials science. The rapid development of artificial intelligence will undoubtedly unlock enormous potential in the future of materials science, which will in turn indirectly drive the advancement of human civilization.