Command Palette
Search for a command to run...
Trained With Fewer Than 100,000 Structured Data Points, the Swiss Federal Institute of Technology in Lausanne (EPFL) Has Proposed PET-MAD, Achieving Atomic Simulation Accuracy Comparable to Professional models.
From semiconductor materials to active drug molecules, electronic structure remains key to understanding performance. First-principles calculations, based on quantum mechanics, can accurately predict the structure, stability, and function of matter, and have driven rapid progress in materials design and drug development. However, the computational complexity increases dramatically with system size.Even top-of-the-line supercomputers struggle to simulate complex processes such as protein folding and catalytic reactions over long timescales.This has created a situation where "the mechanism can be described, but it is difficult to calculate."
As traditional methods approach their computational limits, machine learning offers a new path for atomic-scale simulations. Machine learning of interatomic potentials, trained on quantum mechanical data, maps complex electronic structures into efficient predictive models, reducing computational costs by several orders of magnitude while maintaining near-first-principles accuracy. Over the past two decades, it has demonstrated significant advantages in systems such as high-temperature alloys and biomacromolecules, overcoming the shortcomings of insufficient accuracy in empirical force fields and the prohibitively high cost of first-principles methods. However,Early models were mostly designed for single systems, requiring repeated generation of training data and refitting of parameters, which made cross-system research quite difficult.
Therefore, generalization has become a key direction for further development. In recent years, various general models adaptable to different elements and chemical environments have emerged.However, due to insufficient training data and inconsistent evaluation standards, its overall performance remains unstable.
Against this backdrop, the PET-MAD model proposed by the Swiss Federal Institute of Technology in Lausanne (EPFL) provides new samples.Leveraging a dataset covering a wide range of atomic diversity and a network architecture based on the Point Edge Transformer, it achieves accuracy comparable to PET-Bespoke dedicated models while using far fewer training samples than traditional models.This provides a powerful example for the development of atomic simulation towards greater efficiency and wider applicability.
The relevant research findings, titled "PET-MAD as a lightweight universal interatomic potential for advanced materials modeling," have been published in Nature Communications.

Paper address:
https://www.nature.com/articles/s41467-025-65662-7
Follow our official WeChat account and reply "PET-MAD" in the background to get the full PDF.
More AI frontier papers:
https://hyper.ai/papers
MAD dataset: Contains 85 elements and nearly 100,000 structures
The MAD dataset was constructed to provide a high-quality data foundation for training inter-atomic potentials (MLIPs) in general machine learning, which features broad chemical diversity, strict data consistency, and good usability.It contains 85 elements with atomic numbers from 1 to 86 (excluding astatine), totaling 95,595 structures.It is systematically divided into 8 subsets based on its chemical and structural characteristics, which broadly cover bulk crystals, surfaces, clusters, two-dimensional materials and molecular systems.
The core of the dataset comes from a subset of the Materials Cloud 3D database, specifically the MC3D subset.It contains 33,596 individual phase crystal structures. Based on this, an MC3D-rattled subset (30,044 structures) was generated by perturbing the atomic coordinates with Gaussian noise, and an MC3D-random subset (2,800 structures) was constructed by randomly rearranging the types of atoms in the crystal. Both are specifically designed to enhance coverage of extreme or non-equilibrium distortion configurations.
To meet the simulation requirements of surfaces and low-dimensional systems, the dataset includes the MC3D-surface subset (5,589 low-index crystal plane models) and the MC3D-cluster subset (9,071 nanoclusters) obtained from bulk phase cutting, and the MC2D subset (2,676 two-dimensional crystals) is imported from the Materials Cloud 2D database. For molecular systems, it is supported by two subsets: SHIFTML-molcrys (8,578 molecular crystals) and SHIFTML-molfrags (3,241 molecular fragments), all of which are derived from authoritative databases to ensure the reliability of the samples.
Regarding data generation strategies,MAD employs a combination of direct reuse and structural modification. Four subsets—MC3D, MC2D, SHIFTML-molcrys, and SHIFTML-molfrags—directly utilize published structures but recalculate energies and forces using identical DFT parameters, fundamentally eliminating systematic biases between different sources. The remaining subsets, through physically driven modifications to the basic structure (such as adding noise, rearranging atoms, and cutting surfaces), directionally expand the diversity of configurations.
PET-MAD: Architecture and Performance Highlights of a Universal Interatomic Potential Model
The PET-MAD model is trained on an architecture optimized by Pareto Frontier.The architecture consists of two message passing layers, each equipped with two Transformer sublayers, using 256-dimensional token representations, an 8-head multi-head attention mechanism, and finally outputting the result through a fully connected layer with 512 neurons.To improve the stability of the training process, before formal training, the system will fit a linear model and subtract the baseline contribution of energy related to atomic composition.
The entire training was performed within the PyTorch framework using the Metatrain package. The computational tasks were distributed across eight NVIDIA H100 GPUs, with a batch size of 24 architectures per GPU. A total of 1,500 epochs were trained, taking approximately 40 hours. The optimization process employed the Adam optimizer, with an initial learning rate of 10⁻⁴, halved every 250 epochs. The loss function consisted of the root mean square error of the energy and force predictions, with the energy term's weight scaled to 0.1.
After training, the model achieved an average absolute error of 7.3 meV/atom for energy and 43.2 meV/Å for force on the training set, and 14.7 meV/atom for energy and 172.2 meV/Å for force on the validation set, demonstrating excellent fitting accuracy.
To enable the general model to better adapt to specific chemical systems,PET-MAD introduces low-rank adaptation (LoRA) fine-tuning technology.Unlike traditional full-parameter fine-tuning, which can easily lead to "catastrophic forgetting" (i.e., loss of existing general knowledge), LoRA freezes all weights of the base model, injecting only two pairs of trainable low-rank matrices into each attention module, and adjusting their influence through a scaling parameter, thereby achieving the goal of "precise improvement without loss of generality." In this study, the rank of LoRA is fixed at 8, and the scaling parameter is set to 0.5.
Experiments show thatIn scenarios with limited data, models fine-tuned with LoRA consistently outperform dedicated models trained from scratch for this system.Even in complex systems such as barium titanate, fine-tuned models can produce physical observables comparable to dedicated models while maintaining general accuracy, and are therefore recommended as an optimization scheme for specific applications.
For general-purpose models, reliable error estimation is crucial. PET-MAD quantifies uncertainty through the Last Layer Prediction Rigidity (LLPR) method. This method estimates the posterior error of new predictions by analyzing the covariance of the model's last layer of hidden features on the training set, incurring almost no additional computational cost. More valuablely, LLPR allows sampling of a finite number of last-layer weights to form a lightweight "shallow ensemble," thereby transferring uncertainty to the calculation of complex derived quantities such as free energy and phonon dispersion curves. This method has a significantly lower computational cost than traditional ensemble methods, making it well-suited for the practical needs of large-scale atomic simulations.
also,PET-MAD offers two force prediction modes: one is based on automatic differentiation (backpropagation) to deduce the force from energy analysis, which strictly satisfies energy conservation; the other is to predict the force directly from atomic coordinates through a separate neural network module, which can improve the inference speed by 2-3 times, but may introduce a small amount of energy non-conservation.By designing a dedicated multi-time-step integrator, the research team effectively avoided the sampling errors that might be caused by the latter mode, thus ensuring both computational efficiency and the reliability of the dynamic simulation.
Both benchmark and practical advantages: Achieving the accuracy of a dedicated model with minimal proprietary data.
To comprehensively evaluate the performance of PET-MAD, the research team first compared it with mainstream model systems such as MACE-MP-0 and Orb-v2 in authoritative benchmark tests such as Matbench Discovery. To ensure fairness, the researchers recalculated the benchmark subset using PET-MAD-compatible DFT parameters to eliminate bias caused by differences in calculation methods.
The test results are shown in the table below.PET-MAD, trained with fewer than 100,000 structures (1-3 orders of magnitude less data than similar models), and with a moderate number of 2.8 million parameters (far lower than Orb-v2's 25 million and MACE-MP-0-L's 15.8 million), outperforms all comparable models on molecular datasets (SPICE, MD22).It is comparable to state-of-the-art models such as MatterSim-5M in inorganic datasets, and its advantages are more significant on datasets with highly distorted configurations, with some models having an error up to 50 times that of MatterSim-5M.
However, benchmark tests are insufficient to fully verify the reliability of the model in complex scenarios. Therefore, the research team selected six diverse and challenging application cases to compare the performance of PET-MAD, its LoRA fine-tuning model, and the PET-Bespoke dedicated model.
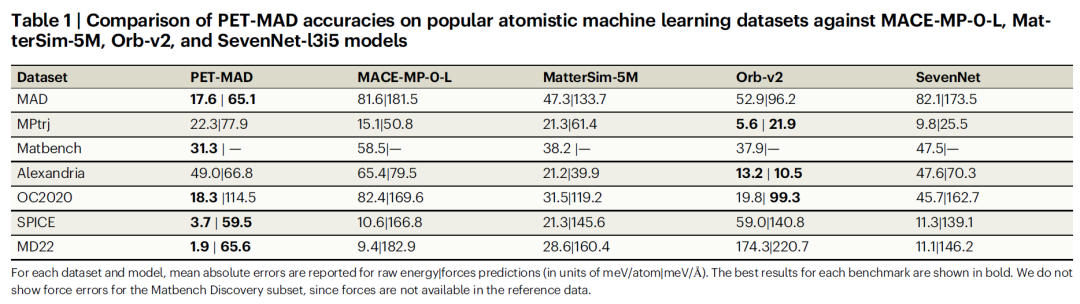
1. Solid electrolyte: Lithium thiophosphate (LPS)
The study focuses on its key performance characteristic of ionic conductivity. As shown in the figure below, although the verification errors of PET-MAD in terms of energy and force (4.9 meV/atom, 163.9 meV/Å) are higher than those of the PET-Bespoke dedicated model (1.2 meV/atom, 35.6 meV/Å).However, the conductivity predicted by the molecular dynamics simulations driven by it is in high agreement with the results of the PET-Bespoke dedicated model and the fine-tuned model over a wide temperature range.It accurately captured the effect of structural motifs (PS₄ tetrahedral rotation) on ion transport, with only a slight overestimation of the phase transition temperature of the γ-phase.
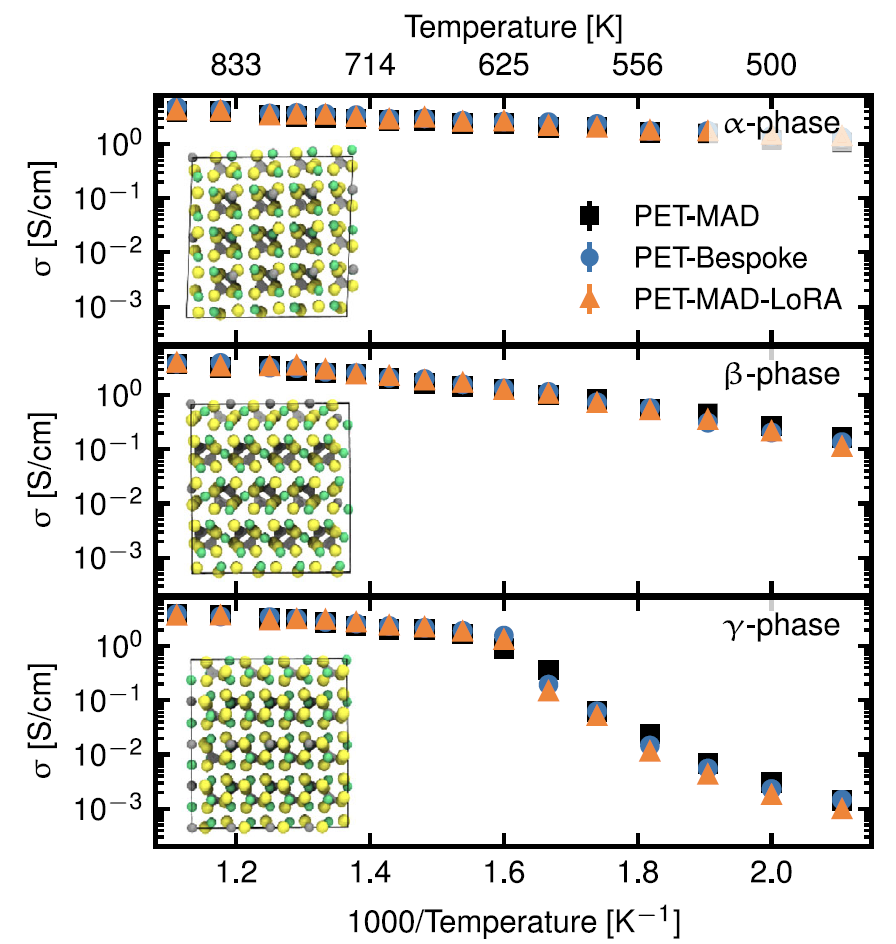
2. Semiconductor material: Gallium arsenide (GaAs)
The results of calculating its melting point using the interface pinning method are shown in the figure below. The LoRA-adjusted PET-MAD is in perfect agreement with the prediction results of the PET-Bespoke dedicated model (1169±3 K vs 1169±4 K).
Although the predicted value of the pre-trained general model (1111±72 K) is slightly lower, its error is much smaller than the inherent deviation of the DFT functional itself from the experimental value (1511 K).This case also highlights the value of PET-MAD's built-in LLPR uncertainty quantification function, which can assess and transmit errors at low cost, providing a reference for the credibility of results.
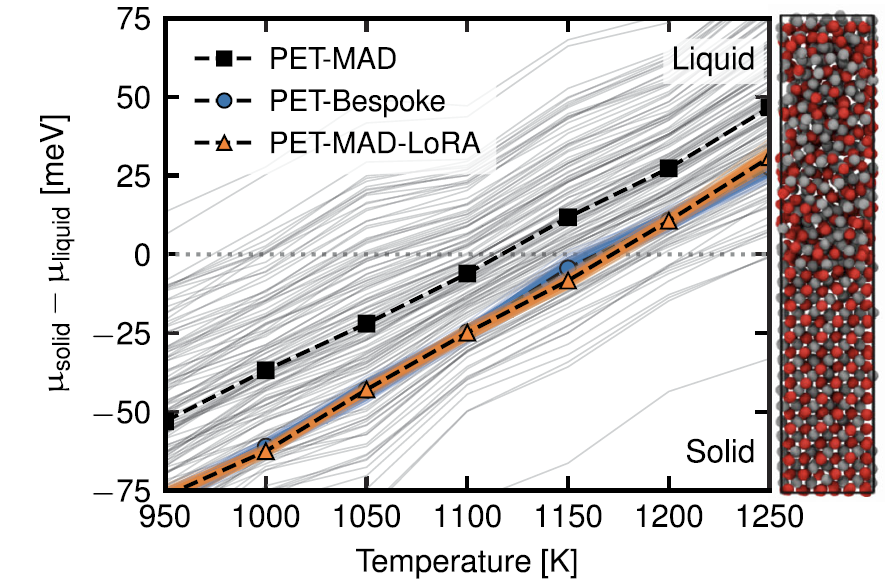
3. High-entropy alloys: CoCrFeMnNi
Regarding its surface elemental segregation behavior, the study found, as shown in the figure below, that the pre-trained PET-MAD can accurately reproduce the segregation characteristics of "nickel-rich surface and depleted other elements" without special training for this system, which is in quantitative agreement with the reference study.
Although its energy error (25.8 meV/atom) is higher than that of the PET-Bespoke dedicated model (14.6 meV/atom).However, the latter may be at risk of overfitting due to limited training data (only 2000 structures), while PET-MAD shows better robustness.
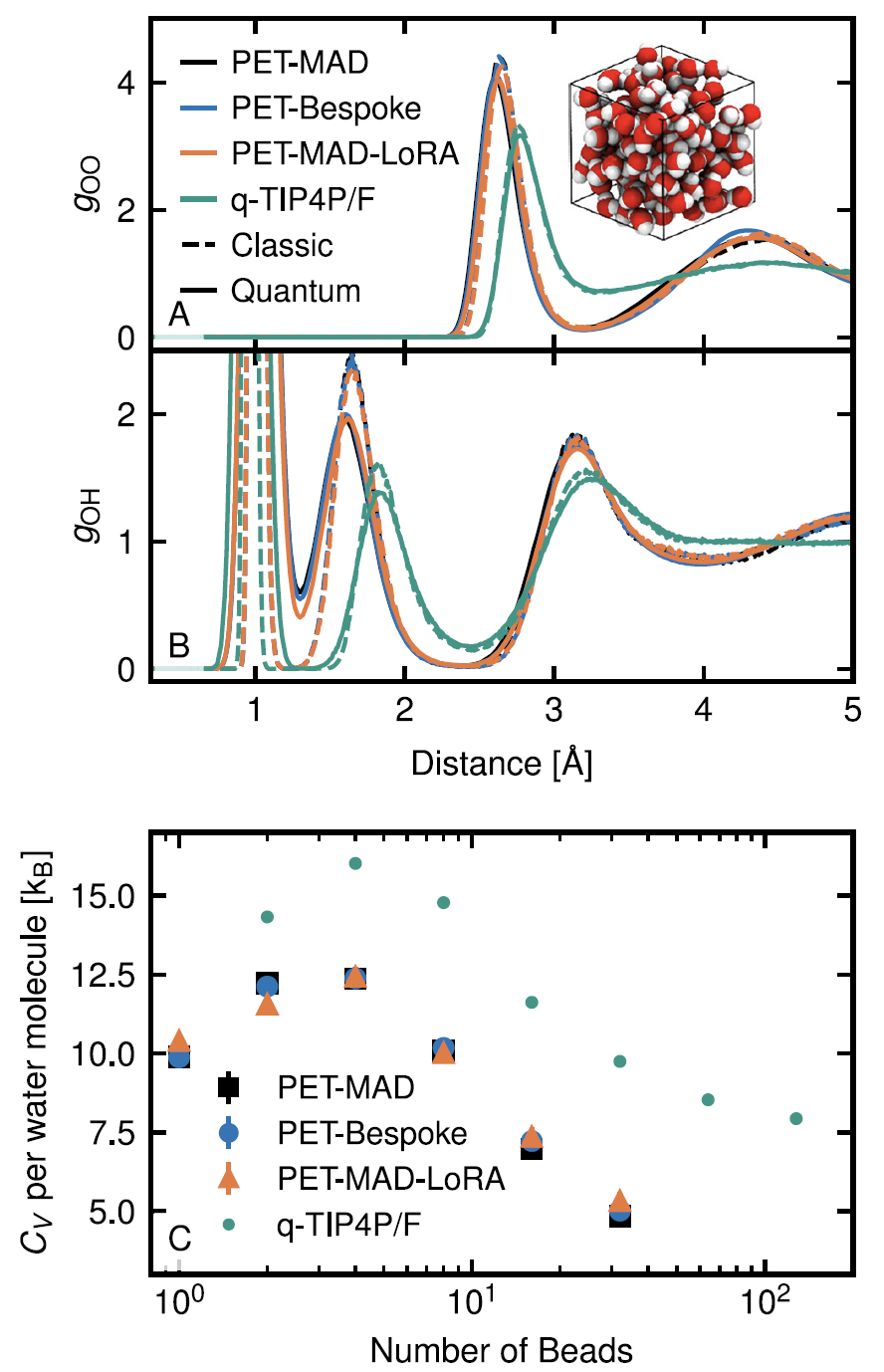
4. Liquid water and the quantum nuclear effect
Through path integral molecular dynamics simulations, PET-MAD successfully characterized the structural ordering and proton delocalization phenomena caused by hydrogen atom quantum fluctuations. Although limited by the GGA functional accuracy it has learned, the simulated system exhibits "supercooled liquid" characteristics at room temperature, but it performs well in calculating the radial distribution function and heat capacity.The results are in high agreement with the PET-Bespoke dedicated model, and its efficiency makes such computationally intensive quantum simulations more feasible.
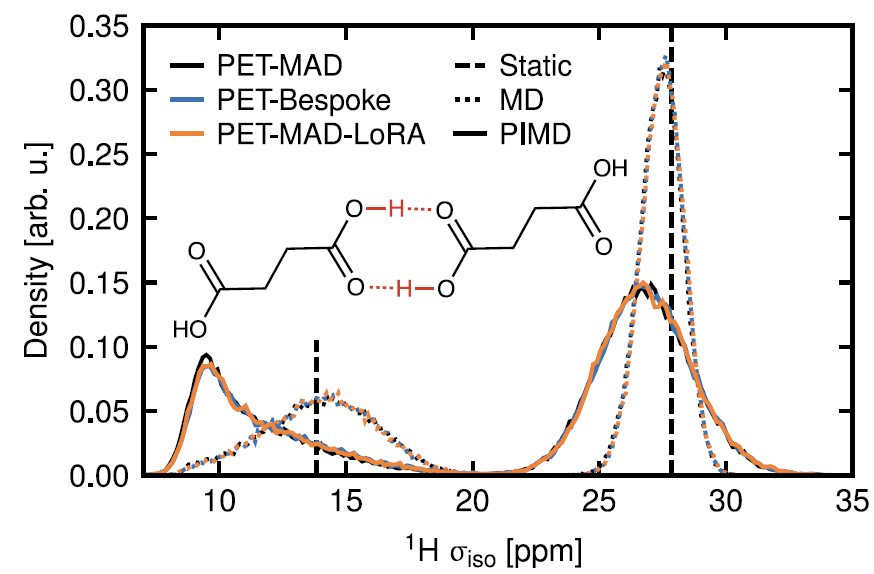
5. Molecular Crystals: NMR Study of Succinic Acid
PET-MAD-driven kinetic simulations combined with a chemical shielding model successfully revealed the influence of nuclear quantum motion on the distribution of the ¹H NMR shielding constant. As shown in the figure below, the shielding distribution of hydrogen-bonded protons significantly broadens and shifts downward under quantum sampling.This is consistent with the conclusions of the PET-Bespoke dedicated model trained on high-precision DFT, demonstrating the potential of PET-MAD in predicting complex functional characteristics.
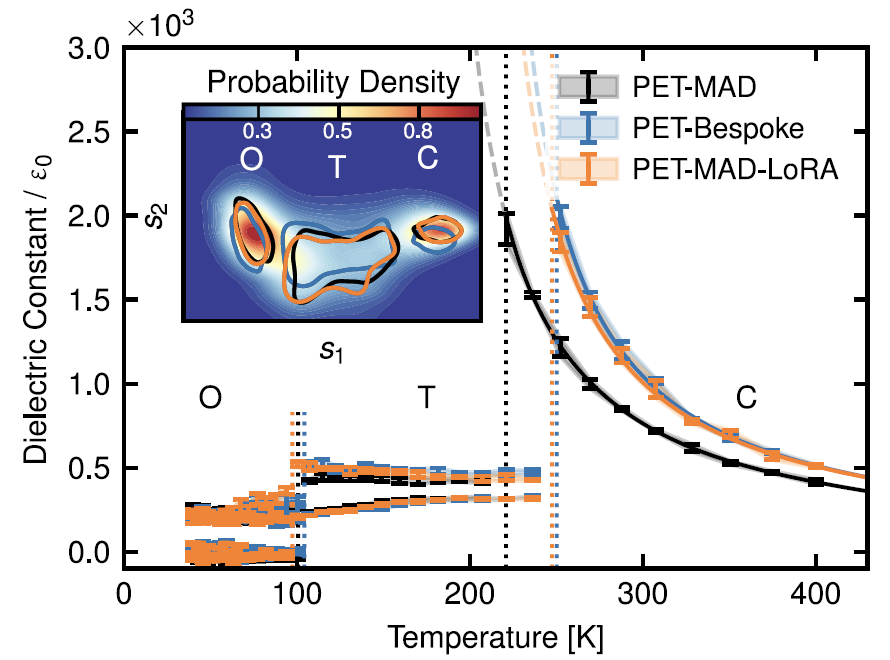
6. Ferroelectric materials: Barium titanate (BaTiO₃)
As shown in the figure below, in flexible cell simulations within the 40-400K temperature range, PET-MAD accurately reproduced the characteristic phase transition sequence of the material from rhombohedral to orthorhombic to tetragonal to cubic. Although limited by DFT...The predicted phase transition temperature is lower than the experimental value, but its deviation from the result of the PET-Bespoke dedicated model is less than 30K.In the calculation of static dielectric constant, it also correctly predicted the trend of high dielectric constant in cubic phase and low dielectric constant in ferroelectric phase.
Both benchmark testing and multivariate system applications demonstrate that...PET-MAD can achieve simulation accuracy close to that of PET-Bespoke dedicated models with very little proprietary data, and reliably predict properties such as ion transport, phase transition and surface reaction.Its LoRA fine-tuning can further approximate the performance of the PET-Bespoke dedicated model, while LLPR error estimation and dual force prediction mode ensure the efficiency and robustness of complex simulations, thus making it a versatile interatomic potential tool with both breadth and depth.
MLIPs: Industry-Academia-Research Integration Drives a Paradigm Shift in Materials Research and Development
With its significant advantages of high precision and high efficiency, the field of MLIPs is increasingly becoming a key bridge connecting the forefront of quantum chemistry theory with industrial engineering applications. In recent years, research has been moving from verifying the feasibility of methods to further expanding their capabilities.
For example, a research team from the University of Cambridge and the Swiss Federal Institute of Technology in Lausanne (EPFL) combined active learning with augmented sampling strategies.This enables the model to autonomously explore key transition state configurations along chemical reaction pathways, thereby achieving accurate predictions of catalyst activity and selectivity without the need for extensive prior reaction data.It effectively overcomes the limitations of existing MLIPs in describing the dynamic processes of chemical reactions.
Paper title: Aedes aegypti Argonaute 2 controls arbovirus infection and host mortality
Paper link:
https://www.nature.com/articles/s41467-023-41370-y
Meanwhile, the systematic evaluation and mechanistic elucidation of existing models are continuously deepening. The University of California team conducted a comprehensive evaluation of mainstream general-purpose MLIPs such as M3GNet, CHGNet, and MACE-MP-0.It covers a range of material properties, including surface energy, defect formation energy, phonon spectrum, and ion migration barrier.The study found that these models generally exhibit a systematic underestimation of energy and force in tasks involving high-energy states or non-equilibrium configurations, which points the way for further improvements in model training strategies and data construction.
Paper Title: Systematic softening in universal machine learning interatomic potentials
Paper link:
https://www.nature.com/articles/s41524-024-01500-6
In summary, the development of the MLIPs field has entered a new stage, moving from algorithmic innovation to problem-driven approaches, and from basic evaluation to in-depth application scenario development. In the future, as more cross-scale modeling challenges are overcome and industry-specific models mature, MLIPs are expected to play a more central role in the digital R&D system, accelerating the mutual empowerment of materials science and industrial innovation.