Command Palette
Search for a command to run...
Cornell University Proposes an Innovative AI Framework to Decode the Chemical Mechanism of Highly Conductive lithium-ion Electrolytes, Achieving a Prediction Success Rate Exceeding 80% for %.

With the rapid expansion of the new energy battery market, especially the widespread application of lithium-ion batteries, solid-state batteries and high-energy-density batteries, the optimization of electrolyte performance has become a key factor determining battery safety, efficiency and lifespan.
Salt-solvent chemistry underpins the electrolyte behavior in most lithium-ion battery systems, determining key properties such as ionic conductivity, viscosity, and chemical stability. However, its rational design is constrained by a vast chemical space encompassing countless combinations and nonlinear structure-performance coupling relationships. Sparse and unevenly distributed experimental data further exacerbates this problem, hindering the generalization ability of models. In recent years, while AI-driven autonomous electrolyte discovery has made some progress,However, existing research still clearly lacks a unified computational paradigm that can maintain inherent interpretability while exploring the vast chemical space covered by large-scale electrolyte formulations.
In this context,A research team from Cornell University has developed a robust, interpretable, and data-efficient framework, SCAN, for modeling and interpreting salt-solvent chemistry.This framework effectively handles long-tailed data and captures the complete spectrum of salt-solvent formulations. Researchers applied SCAN to non-aqueous electrolyte (NAE) systems, achieving a baseline error of 0.372 mS·cm⁻¹ in conductivity prediction, reducing the prediction error by 65.31 TP³T compared to the baseline model.
More importantly, large-scale validation shows thatThe model achieved a prediction success rate of 81.08% for the top-ranked candidate systems.In addition to its predictive capabilities, SCAN reveals the chemical mechanism by introducing gradient decoupling, symbolic regression, and quantum chemical calculations to reveal the influence of molecular flexibility and ion-solvent interactions on conductivity.
The related research findings, titled "A dynamic routing-guided interpretable framework for salt–solvent chemistry," have been published in Nature Computational Science.
Research highlights:
* SCAN fills a key gap in high-performance NAE salt-solvent chemistry research.
Inspired by the atomic-centered potential energy surface model, we developed a multi-feature network (MFNet) with a descriptor-centric dedicated representation and attention mechanism.
* Innovatively, a dynamic routing strategy is introduced into MFNet, enabling the model to accurately predict ionic conductivity over a wide range without altering the original data distribution.

Paper address:
https://www.nature.com/articles/s43588-026-00955-5
Follow our official WeChat account and reply "SCAN" in the background to get the full PDF.
Data dimensions are widely covered
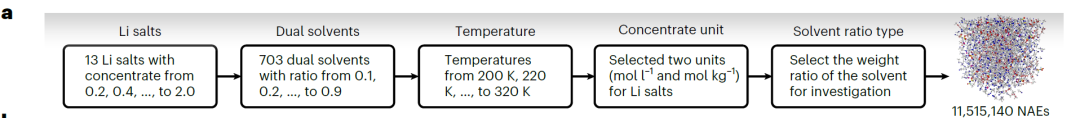
In order to train a high-precision SCAN model,The research team constructed the CALiSol dataset, which includes 13 lithium salts and 38 organic solvents (as shown in the figure below), totaling 13,302 complete data entries.
The data dimensions are extensive, and each data point contains the following:
* Ionic conductivity k: 0–38.1 mS/cm
* Temperature T: 194.15–477.42 K
* Salt concentration c: 0–4 mol/L or mol/kg
* Lithium salt types: LiPF₆, LiTFSI, LiFSI, LiBOB, etc., a total of 13 types.
* Solvent types: including ethylene carbonate (EC), methyl ethyl carbonate (EMC), propionitrile (AN), and many others.
* Mixed Strategy SRT: Molar, Volume, or Mass Ratio
The molecular information of all lithium salts and solvents was converted into three-dimensional molecular coordinates using SMILES sequences, and geometric optimization was performed at the B3LYP/6-31G theoretical level to ensure accurate and reliable molecular structure and electronic properties. Through this method,The dataset provides not only ionic conductivity values, but also molecular characteristics, structural information, and solvation environment for each system.Provide rich inputs for AI models, balancing data integrity and scientific interpretability.
During the construction of the dataset, the research team paid special attention to the problem of long-tailed data (LTD): the number of highly conductive systems is limited, while the conductivity of most systems is low - k < 5 mS·cm⁻¹ for 9,115 NAEs (about 68.5%), and k > 20 mS·cm⁻¹ for only 67 (about 0.5%).
SCAN: Employs MFNet and dynamic routing mechanisms
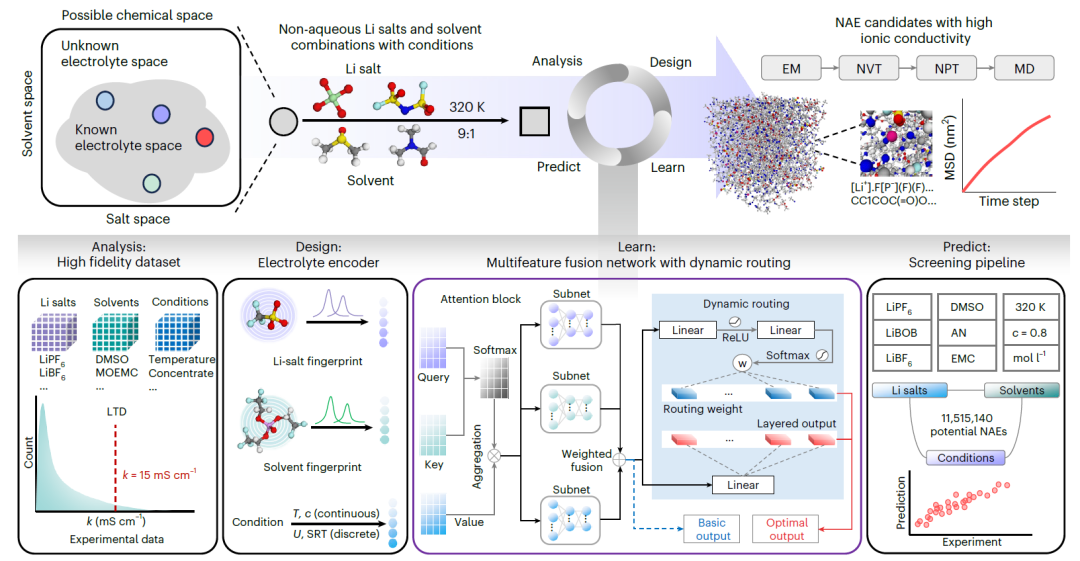
SCAN is a NAE engineering platform guided by dynamic routing. The following diagram illustrates its complete workflow:
Analysis
A high-fidelity experimental dataset was constructed, covering a variety of combinations of lithium salts, solvents, and experimental conditions. Statistical analysis was performed on each variable, including characterization of the long-tailed distribution of k values.
Design
Molecular fingerprints with chemical information were designed for lithium salts and solvents respectively, and combined with conditional coding to accurately characterize salt-solvent-condition combinations.
Learn
A custom multi-feature fusion network (MFNet) was developed to predict the k value of NAEs. Independent attention modules were constructed by embedding query, key, and value to handle the representations of salt, solvent, and condition, respectively, and then fused using a fully connected neural network. In addition, a dynamic routing strategy was introduced to improve model performance and robustness.
Predict
A high-throughput screening pipeline was constructed to identify potential NAEs from over ten million salt-solvent combinations. Candidate systems with high predicted k values were further validated using molecular dynamics (MD) simulations, including energy minimization (EM), canonical ensemble (NVT), isothermal-isobaric ensemble (NPT), MD production simulations, mean square displacement (MSD) analysis, and radial distribution function (RDF) calculations.
MFNet and the dynamic routing mechanism are the core of the entire framework, and the specific mechanisms are as follows:
MFNet: Multichannel Self-Attention Network
Inspired by the atomic central potential surface model, the MFNet (Molecular Feature Network) framework divides the network into three independent subnets, each handling different functions:
* Lithium Salt Subnet: In the CALiSol dataset, each NAE contains only one type of lithium salt, so its descriptor vector is directly input into the lithium salt subnet.
* Solvent Subnet: Since some data points involve multiple solvents (e.g., PC and AN mixed in a 0.9:0.1 ratio), the average value of these solvent descriptors is calculated as input to reflect the overall solvent environment.
* Conditional Subnet: Handles experimental conditions such as temperature and concentration.
Following the self-attention module, two fully connected hidden layers are used for progressive projection and non-linear transformation of the input, generating high-dimensional features to enhance the expressiveness of subsequent processing. The ReLU non-linear activation function is used between layers to further improve the model's representational power.
The final network architecture is as follows: a lithium salt subnet (14–16–16), a solvent subnet (14–16–16), and a conditional subnet (6–16–16). Potential outputs with intrinsic dimensions (feature dimensions <128) are fed into a weighted fusion module to compute weighted outputs while preserving global dependency information of the input features. Since the encoding of lithium salt, solvent, and conditional features is processed independently by three subnets, single-head self-attention (SHA) can be used to lightweightly process small-scale feature data (14, 14, and 6 dimensions, respectively), making it well-suited for tasks with low embedding dimensions.
Dynamic routing mechanism: solving the problem of long-tail data
In a long-tailed distribution, data samples are mainly concentrated in the "head region," while samples in the "tail region" are scarce. However, traditional models often overfit to the head samples and ignore the tail region, resulting in insufficient exploration of the critical but scarce chemical space. To address this issue, this study introduces a dynamic routing strategy into MFNet. Unlike the standard architecture that treats all samples equally,Dynamic routing learns a soft gating mechanism to adaptively assign different representation capabilities to different layers based on the layer input.As shown below:

This mechanism enables rare samples to activate different routing paths and facilitate conditional computation, thereby improving generalization ability for low-frequency categories. Its two key features are:
* Input-dependent routing weights: that is, selecting a feature subspace for each sample without modifying the original loss function or data distribution;
* Category-adaptive feature decoupling: This involves explicitly modeling the difference between the dominant and tail categories, demonstrating that dynamic routing provides a more flexible and interpretable LTD solution than simple static loss reshaping.
Interpretability: GBA and Symbolic Regression
SCAN provides chemical interpretability while accurately predicting ionic conductivity:
* GBA (Gradient-based Attribution):Because the SCAN model employs a three-parallel attention neural network framework, its decision-making process is more complex to visualize compared to tree models. To identify the key chemical factors influencing k, the GBA method was used to evaluate feature importance—calculating the gradient contribution of each input feature to the model output, and identifying the most critical lithium salt, solvent, and conditional features.
* Symbolic Regression:To discover an interpretable functional relationship between key Li salt, solvent, and condition information and kkk, a symbolic regression method based on PySRRegressor was used.
The SCAN model consistently outperforms all baseline models.
To comprehensively evaluate the performance of the SCAN framework using MFNet and dynamic routing, researchers constructed four baseline models based on MFNet.None of them contain dynamic routing: (1) Basic MFNet model: B1 (unprocessed descriptor) and B2(Descriptor scaled by maximum value); (2) MFNet model combining minority class oversampling technique SMOTE: S1–S18(3) MFNet model combining K-nearest neighbor undersampling technique (KUTE): U1–U9(4) MFNet model combining SMOTE and KUTE: SU1–SU9 .
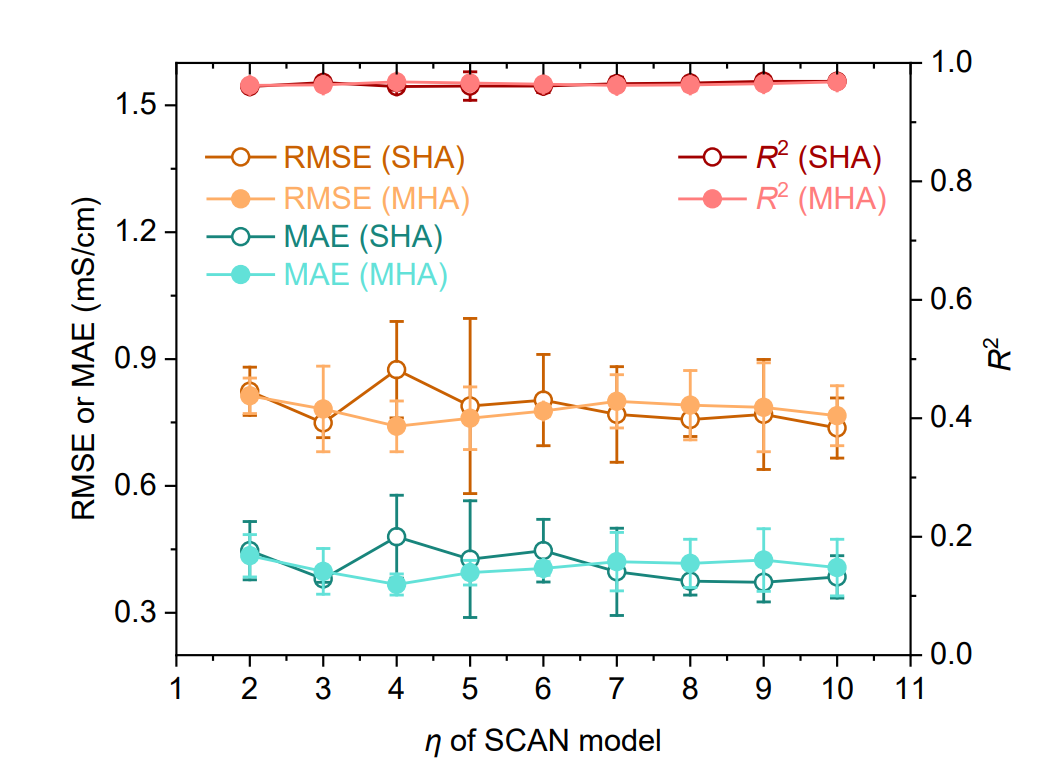
Simultaneously, Transformer-based models, Graph Neural Networks (GNNs), Support Vector Regression (SVR), and Extreme Gradient Boosting (XGBoost) were used as baseline comparisons, as shown in the figure below. Furthermore, SCAN models employing single-head attention (SHA) or multi-head attention (MHA) structures and different numbers of dynamic routing layers (η = 2–10) were implemented to verify model robustness.
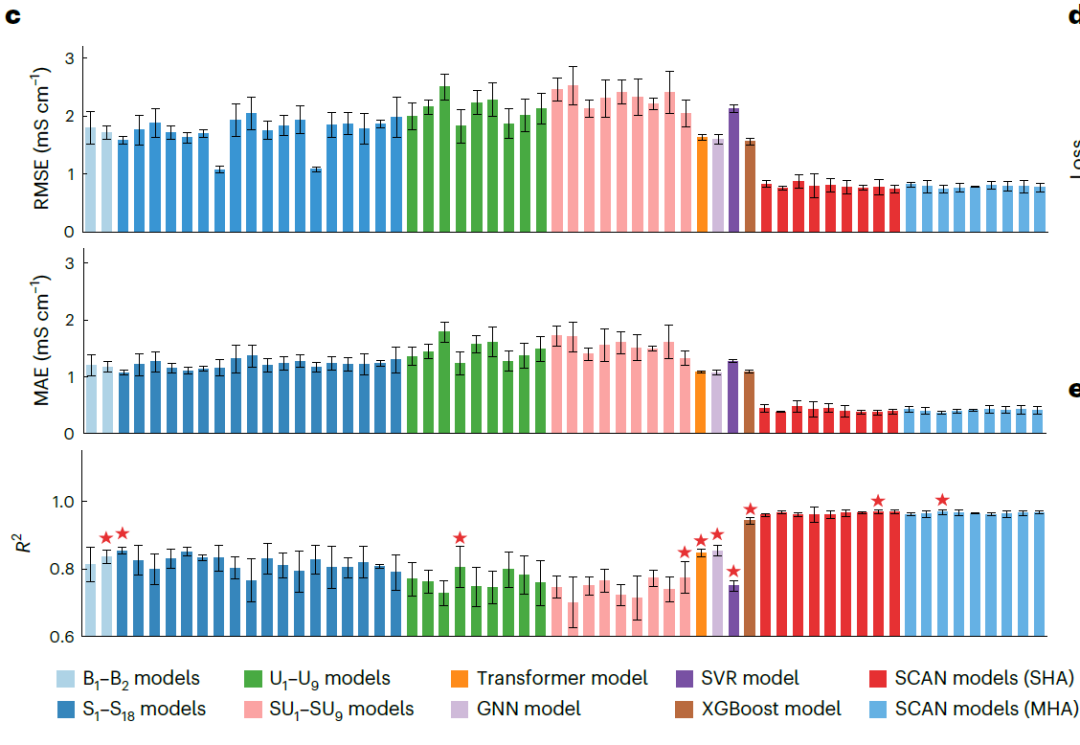
NAE performance evaluation
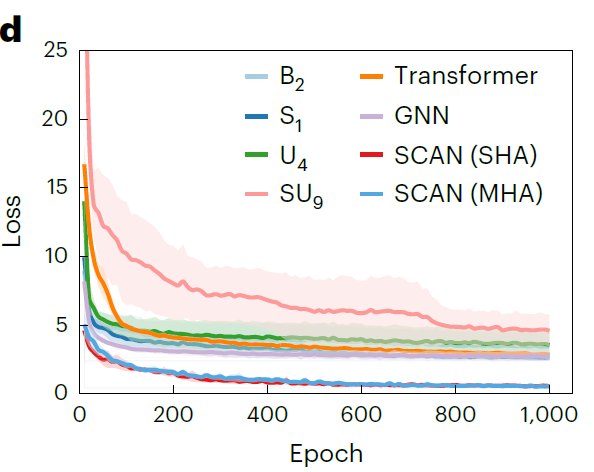
The results shown in the figure below demonstrate that the SCAN model using SHA (η = 9) consistently outperforms all baseline models, exhibiting significantly lower prediction errors (RMSE 0.769 mS·cm⁻¹, MAE 0.372 mS·cm⁻¹) and a higher R² (0.969). It is worth noting that...Compared to the best-performing baseline GNN in MAE (1.072 mS·cm⁻¹), the error was reduced by 65.31 TP3T.The SCAN performance using MHA (η = 4) is comparable to that of the SHA version, indicating that MFNet has good robustness in integration with dynamic routing.
also,Researchers plotted the validation loss curves of SCAN and a representative baseline model over 1,000 training epochs.As shown in the figure below, from the beginning of training, the prediction error of SCAN is significantly lower than that of the baseline model, with an initial loss of only 4.59 mS·cm⁻¹, and its validation loss continues to decrease and converges to an even lower level, indicating that it has stronger stability and generalization ability.
High-throughput NAE screening capability
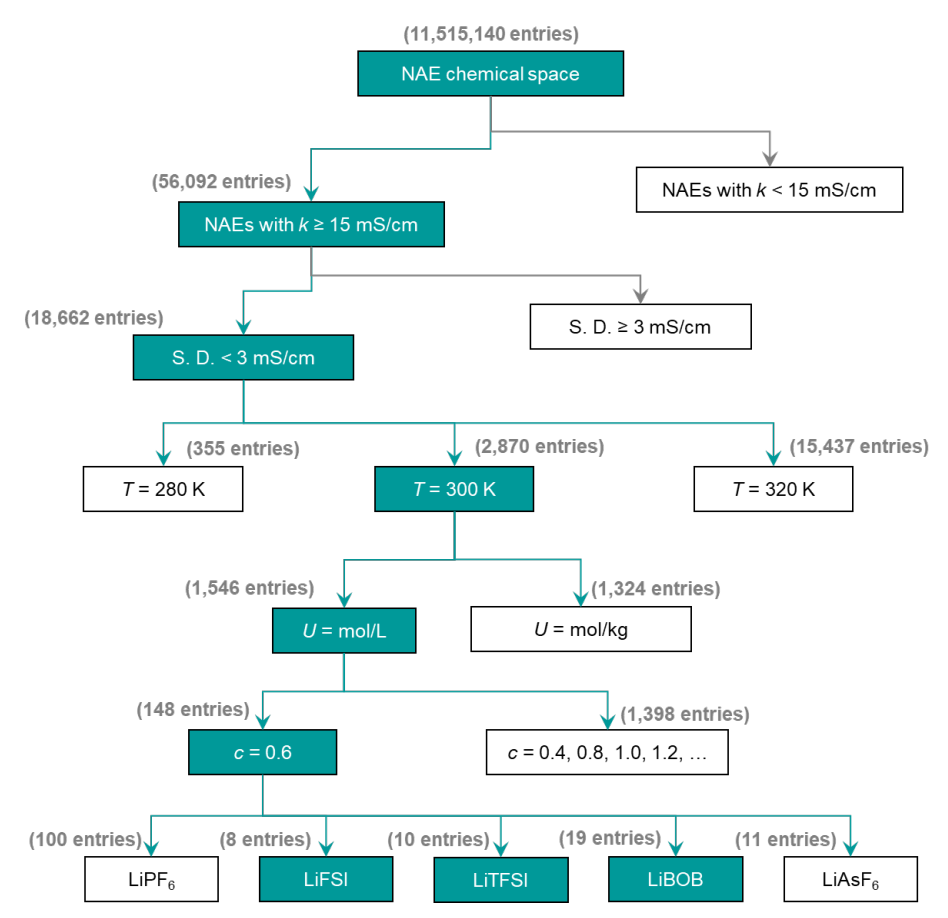
High-precision scanning can efficiently explore the extended chemical space of NAEs. To this end, researchers have developed a high-throughput screening process (as shown in the figure below) designed to identify high-k NAEs.Based on this, 11,515,140 potential dual-solvent NAEs were generated.
By leveraging the predictive power of SCAN, 56,092 NAEs with k ≥ 15 mS·cm⁻¹ were quickly identified, of which 18,662 had low prediction uncertainty (<3 mS·cm⁻¹). This process significantly reduces the computational and cost burden of molecular dynamics simulations and experiments.
Validation of candidate non-aqueous electrolytes (NAEs)
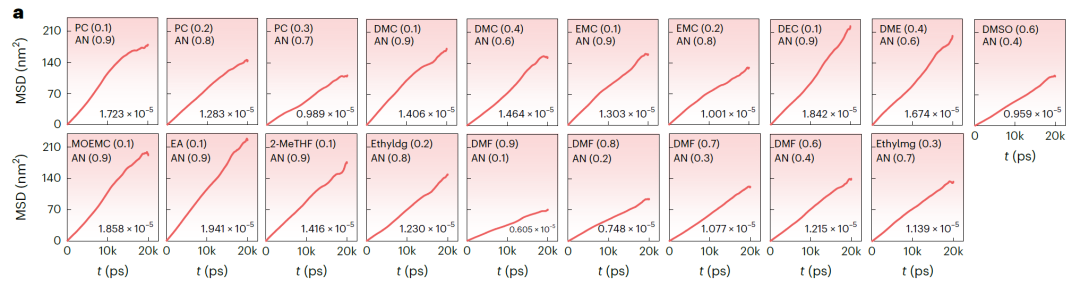
To rigorously validate the predictions, the researchers performed molecular dynamics (MD) simulations to obtain the k value. Based on temperature (T), concentration (c), and actual conditions, they narrowed down the MD validation scope to NAE candidate systems based on LiFSI, LiTFSI, and LiBOB, ultimately selecting 37 promising systems for detailed study, as shown in the figure below:
The average computation time for each system is approximately 10 hours (36,355.12 seconds), and the estimated cost of simulating 10⁷ candidate systems is approximately 10⁸ GPU hours—far exceeding the practical capabilities of brute-force methodological selection, even under ideal high-performance computing conditions. In comparison,A well-trained SCAN model can complete the entire process from descriptor computation to final prediction in less than 5 seconds for each candidate system, reducing computational costs by more than 7,200 times.This significantly improves scalability and efficiency. This highlights the necessity of the SCAN framework in NAE discovery, whose surrogate model can quickly prioritize high-performance candidates with extremely low computational cost.
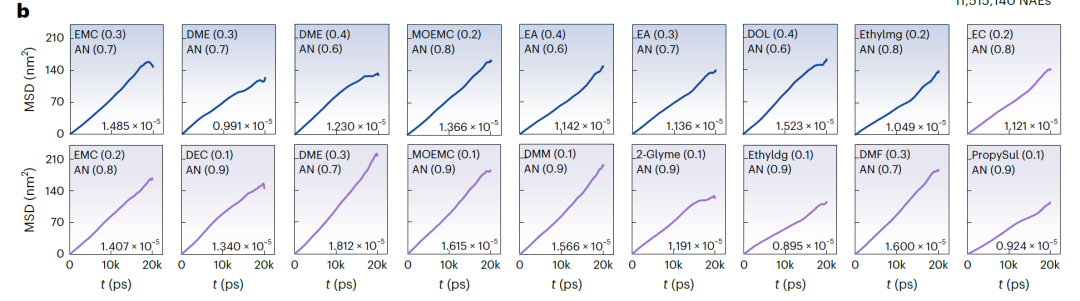
Figure b (NAEs based on LiFSI and LiTFSI) and Figure a (NAEs based on LiBOB) below show the mean square displacement (MSD) over time and the corresponding diffusion coefficient obtained from MD simulations. The MSD of all systems shows a stable linear increase, indicating that the simulation is stable and convergent. The k values predicted by SCAN are highly consistent with the MD simulation results, with an average deviation of only 3.198 mS·cm⁻¹ and a maximum deviation of 7.342 mS·cm⁻¹. In the validation systems: for 25 systems with k > 15 mS·cm⁻¹, the success rate was 67.571TP⁻¹. For systems with predicted k > 14 mS·cm⁻¹, the validation success rate increased to 81.081TP⁻¹.
Artificial intelligence is reshaping the fundamental paradigm of battery research and development.
Batteries, as a core component in key applications such as consumer electronics, electric vehicles, and grid energy storage systems, are driving the global sustainable energy transition. Improving energy and power density, extending cycle life, enhancing safety, and reducing manufacturing costs are the core goals of battery research and development. The key to achieving these goals lies in a deep understanding of the electrochemical mechanisms within batteries, specifically including electrochemical interface reactions and stabilization mechanisms, the coupling and transport of electrons and ions, and the energy storage mechanisms of next-generation electrode materials.
From a broader perspective of technological evolution, the "data-driven + interpretable modeling" paradigm represented by the SCAN framework is becoming an important component of the next-generation battery R&D system. In the past few decades, innovation in battery material systems has mainly relied on experience-driven and trial-and-error experiments, resulting in long development cycles and high costs. However, with the integration of machine learning and high-throughput computing, battery R&D is becoming increasingly efficient.
For example,Wen Yan and Sheng Gong, among others, from ByteDance's Seed team, developed a unified framework for electrolyte formulation design that integrates forward prediction models with reverse generation methods.Researchers extensively collected literature data on single molecules (over 240,000) and molecular mixtures (over 10,000) with property tags, thus broadly covering the electrolyte design space. By further incorporating over 100,000 molecular mixture data from molecular dynamics simulations, they were able to train an accurate machine learning model that can predict not only conductivity but also melting structures related to the interfacial stability of lithium metal batteries.
Paper title: A unified predictive and generative solution for liquid electrolyte formulation
Paper link:
https://www.nature.com/articles/s42256-025-01173-w
Overall, artificial intelligence is reshaping the underlying paradigm of battery research and development. With the deep integration of models and experimental systems, the speed of discovery and innovation density of battery materials are expected to enter a new accelerated cycle.
References:
https://www.nature.com/articles/s43588-026-00955-5
https://phys.org/news/2026-02-ai-framework-reveals-chemistry-high.html
https://static-content.springer.com/esm/art%3A10.1038%2Fs43588-026-00955-5/MediaObjects/43588_2026_955_MOESM1_ESM.pdf
https://www.eet-china.com/mp/a471613.html